| Water mixed with sodium & chloride ions (README) | ||||||
")
|
|
")
")
|
|

|
|
")
|
| spce.lt (water model) | ions.lt | system.lt | run.in.npt | |||
| Build Using: |
moltemplate.sh system.lt |
|||||
| Run Using: |
lmp_mpi -i run.in.npt |
|||||
| Mixture of two small organic molecules using the OPLSAA force field (README) | ||||||

|
|

|
|

|
|
")
|
|
ethylene.lt
oplsaa2024.lt |
benzene.lt | system.lt | run.in.npt | |||
| Build Using: |
moltemplate.sh system.lt -report-duplicates bytype __ |
|||||
| Run Using: |
lmp_mpi -i run.in.nvt |
|||||
| Building a short polymer ("butane") using the COMPASS force field (README) | ||||||


|
|

|
|

|
|
")
|
|
ch2.lt
ch3.lt compass_published.lt |
butane.lt | system.lt |
run.in.npt
run.in.nvt |
|||
| Build Using: |
moltemplate.sh system.lt |
|||||
| Run Using: |
lmp_mpi -i run.in.npt lmp_mpi -i run.in.nvt |
|||||
| Building a NIPAM polymer in water with ions using the OPLSAA force field (README) | ||||||

|
|

|
|

|
|
")
|
|
NIPAM.lt (monomer)
oplsaa2024.lt |
NIPAM_polymer_10mer.lt |
spce.lt
ca.lt cl.lt |
system.lt |
run.in.min
run.in.npt run.in.nvt |
||
| Build Using: |
moltemplate.sh system.lt |
|||||
| Run Using: |
lmp_mpi -i run.in.min lmp_mpi -i run.in.npt lmp_mpi -i run.in.nvt |
|||||
| Building a long alkane chain (using the LOPLS force field) (README) | ||||||

|
|

|
|

|
|

|
|
ch3.lt
loplsaa2024.lt |
ch2.lt |
alkane50.lt
system.lt (Note: more complex shapes are possible) |
README.md
run.in.min run.in.nvt |
|||
| Build Using: |
moltemplate.sh system.lt |
|||||
| Run Using: |
lmp_mpi -i run.in.min
lmp_mpi -i run.in.nvt
|
|||||
| Carbon-Nanotube capillary (all-atom, explicit water) (README) | ||||||

|
|
 (reader's comment:
atoms near junction should be adjusted.
Chiral nanotubes need special treatment.)
(reader's comment:
atoms near junction should be adjusted.
Chiral nanotubes need special treatment.)
|
|

|
|
 ....
....

|
|
graphene.lt |
nanotube.lt, graphene_walls.lt |
spce.lt, water_box.lt |
system.lt, run.in.nvt, (video) |
|||
| Build Using: |
moltemplate.sh system.lt |
|||||
| Run Using: |
lmp_mpi -i run.in.nvt |
|||||
| Aggregation of Simple Toy Polymers (coarse grained) | ||||||
")
|
|

(Note: more complex shapes are possible) |
|

|
|

|
|
monomer.lt
forcefield.lt |
polymer.lt | system.lt | run.in.nvt | |||
| Build Using: |
moltemplate.sh -atomstyle "full" system.lt |
|||||
| Run Using: |
lmp_mpi -i run.in.nvt |
|||||
| Translocation of a short polymer through a pore in explicit LJ solvent (README) | ||||||
| Requirements: | This example requires that LAMMPS is built with the optional RIGID package. | |||||
")
|
|
")
|
|

|
|
")
|
|
solvent_single.lt, solvent.lt |
wall_single.lt, walls.lt |
monomer.lt, polymer.lt, polymer_forcefield.lt |
system.lt, run.in.npt, (video) |
|||
| Build Using: |
moltemplate.sh system.lt |
|||||
| Run Using: |
lmp_mpi -i run.in.npt |
|||||
| Coarse-grained lipid bilayer using the MARTINI force field and PACKMOL (README) | ||||||
| Requirements: | This example uses PACKMOL (For an alternative method, see this example.) | |||||

 water model")
| |

|
|

|
|

|
|
README.md
DPPC.lt water.lt system.lt martini.lt |
README_packmol.txt
README_moltemplate.txt DPPC.xyz water.xyz mix_lipids+water.inp |
run.in.min
run.in.anneal run.in.nvt |
(video) | |||
| Build Using: |
packmol < mix_lipids+water.inp # (create the "system.xyz" file) moltemplate.sh -xyz system.xyz system.lt |
|||||
| Run Using: |
lmp_mpi -i run.in.min lmp_mpi -i run.in.anneal lmp_mpi -i run.in.nvt |
|||||
| Many-Body force field example: mW solvent + CG hydrocarbon mixture (Many-body force fields can be combined with ordinary, pairwise-additive force fields. README.) | ||||||
| Requirements: | This example requires that LAMMPS is built with the MANYBODY package. | |||||
")
|
|
")
|
|

|
|
")
|
|
README.png
watmw.lt |
cyclododecane.lt, trappe1998.lt |
README.TXT
system.lt |
run.in.npt (video) | |||
| Build Using: |
moltemplate.sh -a "@atom:/WatMW/mW 1" system.lt |
|||||
| Run Using: |
lmp_mpi -i run.in.npt |
|||||
| Coarse-grained membrane protein (README) | ||||||
| Requirements: | This example requires that LAMMPS is built with the optional MISC package, before additional code is added (in that order) | |||||
|
|
|
")
|
|
")
|
|

|
|
CGLipidBr2005.lt, table_int.dat |
1beadProtSci2010.lt, |
system.lt | run.in.npt | |||
| Build Using: |
moltemplate.sh system.lt |
|||||
| Run Using: |
lmp_mpi -i run.in.npt |
|||||
| Vesicle with protein inclusions (README) | ||||||
| Requirements: | This example requires PACKMOL. LAMMPS must be built with the optional MISC package, before additional code is added (in that order) | |||||
|
|
|

|
||||
|
Moltemplate files: README CGLipidBr2005.lt table_int.dat 1beadProtSci2010.lt system.lt |
PACKMOL files: README_pm DPPC.xyz protein.xyz step1_proteins.inp step2_innerlayer.inp step3_outerlayer.inp |
|
LAMMPS files: run.in.min run.in.make_uniform run_T=345K.in This is a complex example requiring hours or days to set up. Please follow the instructions in the README files. |
|||
| Build Using: |
packmol < step1_proteins.inp # requires ~40 minutes packmol < step2_innerlayer.inp # requires ~10 hours packmol < step3_outerlayer.inp # requires 1-3 days (creates "system.xyz" file) moltemplate.sh -xyz system.xyz system.lt |
|||||
| Run Using: |
lmp_mpi -i run.in.min lmp_mpi -i run.in.make_uniform lmp_mpi -i run_T=345K.in |
|||||
|
DNA polymer that follows a curve
(README.md)
WARNING: To create an equilibrium polymer melt, you must use a "soft" potential to allow the polymer(s) to fully relax. |
||||||
| Requirements: | This example requires that LAMMPS is built with the optional MISC package. | |||||
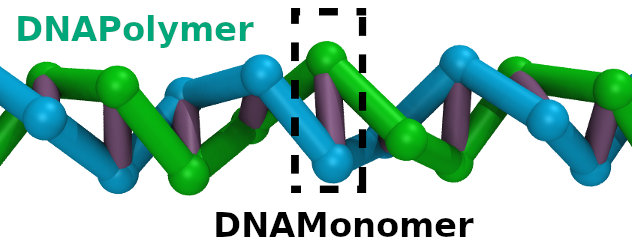
|
|

|
|
")
|
|

|
|
README.md
dna_monomer.lt dna_forcefield.lt |
curve_smooth.txt
(generated by ndmansfield and interpolate_curve.py ) |
dna_polymer.lt
(generated by genpoly_lt.py See below.) |
wall_particle.lt
wall.lt system.lt run.in.min |
|||
| Build Using: |
NMONOMERS=3058; # number of monomers
B=0.996 # length per monomer
Nx=11; Ny=11; Nz=11 # number of lattice sites
# Generate a lattice polymer shape (integer coordinates)
# Note: You must download "ndmansfield" at https://github.com/jewettaij/ndmansfield
./ndmansfield -box $Nx $Ny $Nz -seed 0 -tstart 1 -tsave 1000000 -tstop 1000000 > curve_lattice.txt
# Interpolate and rescale the coordinates:
SCALE=`echo "($NMONOMERS*$B)/($Nx*$Ny*$Nz-1)" | bc -l` #physical curve length / #lattice sites
interpolate_curve.py $NMONOMERS $SCALE < curve_lattice.txt > curve_smooth.txt
# Create a moltemplate file ("dna_polymer.lt") for a polymer with this shape
genpoly_lt.py -helix 102.7797 \
-polymer-name 'DNAPolymer' \
-monomer-name 'DNAMonomer' \
-inherits 'DNAForceField' \
-header 'import "dna_monomer.lt"' \
-bond Backbone a a \
-bond Backbone b b \
-dihedral MajorGroove b b a a 0 1 1 2 \
-dihedral Torsion a a b b 1 0 0 1 \
-padding 20,20,20 \
< curve_smooth.txt > dna_polymer.lt
# (Note: The "system.lt" file contains a link to "dna_polymer.lt".)
# Now run moltemplate to convert "system.lt" into LAMMPS format.
moltemplate.sh system.lt
|
|||||
| Run Using: |
lmp_mpi -i run.in.min #relax the polymer conformation |
|||||
| Ellipsoidal particles (Moltemplate can build systems containing ellipsoids and point-dipoles. README.md) | ||||||
| Requirements: | This example requires that LAMMPS is built with the optional ASPHERE package. | |||||

|
|

|
|
")
|
||
|
README.md
benzene_cg.lt |
system.lt | run.in | ||||
| Build Using: |
moltemplate.sh -atomstyle "atomid atomtype flag density x y z" system.lt
-allow-wildcards -nocheck
|
|||||
| Run Using: |
lmp_mpi -i run.in |
|||||
{kind=link}